
|
 |
 Dr. James Eckman heads Emory's sickle cell disease program, which recently procured a $7.5-million grant from the National Institutes of Health. |
For such a miniscule mistake, the consequences are tragic. Red blood cells carrying the abnormal molecule (hemoglobin S) travel normally through the arterial circulation until they are deoxygenated. "When this happens," says Emory sickle cell expert Dr. James Eckman, "the hemoglobin S molecules often form long, rigid rods, causing the normally Cheerio-shaped cells to stiffen and distort into a sickle shape." These sickle cells have a very hard time moving through the small capillaries. As blood flow slows, he explains, the endothelial cells lining the vessel wall become sticky, attracting the sickle cells like flypaper and causing massive circulatory gridlock. Although these changes are partly reversible through the normal process of reoxygenation, by the time this reversal takes place much damage already has been done. The spleen, acting as policeman, traps and destroys many of the abnormal sickle cells, resulting in rapid turnover of red blood cells and chronic hemolytic anemia. This anemia results in fatigue and a marked susceptibility to infections. Blocked capillaries and veins lead to ischemia in the tissues they supply, causing excruciating pain in bones and muscles as well as progressive organ damage. The course of sickle cell anemia varies considerably. Tremendous advances in detection and treatment mean that most patients now survive to adulthood - many into their 50s and 60s and beyond. Some patients lead fairly normal lives - attending school and work with only occasional pain episodes and slowly progressive organ damage. Others are plagued from an early age with lengthy pain crises (sometimes up to 15 or 20 each year); strokes; bone and joint deterioration; and pulmonary, gallbladder, kidney, or retinal disease - all requiring frequent hospitalization. Until the mid 1970s, 20% to 25% of children born with sickle cell anemia died of overwhelming infections before the age of 5. Although these deaths could easily have been prevented, many times parents and physicians did not realize children had the disease until it was too late. Around the late 1970s, the medical community began to realize that early detection and careful follow-up were the keys to avoiding the high premature death rate from sickle cell disease. |
 
 Red blood cells are normally Cheerio-shaped (left). When they distort into a sickle shape, the result is massive circulatory gridlock, causing excruciating pain and organ damage.  Sickle cell specialist Iris Buchanan with patient Shanna Humphrey. |
 n 1978, Emory recruited Dr. Eckman for the specific purpose of initiating a comprehensive program for sickle cell patients. At the time, there were no organized programs in Georgia for either children or adults with this disease. n 1978, Emory recruited Dr. Eckman for the specific purpose of initiating a comprehensive program for sickle cell patients. At the time, there were no organized programs in Georgia for either children or adults with this disease.
Today, 16 years later, an extensive array of services is available for patients of all ages. Moreover, Emory has now garnered a five-year, $7.5-million grant to establish a National Institutes of Health Comprehensive Sickle Cell Center for clinical and laboratory research. This grant, awarded only every five years, certifies the depth and quality of Emory's program. Last year, when the medical school first submitted its application for this grant, joining 21 other contenders for ten available spaces, its proposal was ranked among the top five. "This is a unique accomplishment," says Dr. Eckman, "considering that no other previous applicant, except in the first round in the early 1970s, has ever even been successful the first time." This national award is only the most recent in a series of grants procured over the years to fund treatment of sickle cell disease. During his first five years here, for example, Dr. Eckman and his colleagues obtained a federal grant from the Maternal and Child Health Program of the Department of Health and Human Services to establish a newborn screening program at Grady Memorial Hospital for sickle cell disease and a basic follow-up for hospital patients. These programs were immediately successful in saving hundreds of lives. A more comprehensive strategy was next on the agenda. Acute problems not requiring hospitalization still were handled at that time in medical and pediatric emergency clinics throughout Atlanta. Patients with uncomplicated pain crises often were placed low on the triage list. Hematologists or generalists provided only fragmented follow-up care for these patients, and continuity of care often was absent. In 1983, a vocal parent-patient group, joined by Emory physicians and Grady Hospital administrators, took their case to the Georgia General Assembly, voicing concern over the level of outpatient care for sickle cell patients. In 1984, the General Assembly responded by providing $550,000 to partially fund a 24-hour, dedicated, comprehensive sickle cell acute care center. This grant, which is still in place, created the Georgia Sickle Cell Center, located on the 15th floor of Grady. The center's mission is to provide tertiary care for all Georgians with sickle cell disease. It currently serves 1,600 patients, 1,061 of whom are treated on a regular basis. Each year, 115 new patients enter the center's program. Sickle cell screening for newborns is now a statewide program, and about 90% of children born with sickle cell anemia now survive to at least age 20. Although the patient population has doubled since the center opened, the number of acute and follow-up visits has remained relatively constant. Many serious complications and hospitalizations are avoided through early intervention, and hospital admissions have decreased significantly, from 215 per 100 patient years for adults in 1985 to 68 per 100 patient years for adults in 1991. |
  One in 400 African-American babies is born with homozygous sickle cell anemia (the most common form); one in 12 carries the sickle cell trait.  Dr. Robert Nerem, an engineer from Georgia Tech who is collaborating with Emory physicians, studies what happens when sickle cells collide with the wall of the blood vessel. |
 esearch is one key to keeping such statistics headed in the right direction. Over the past nine years, the Georgia Sickle Cell Center has initiated clinical studies of analgesic agents for acute pain, transfusion therapy for neurologic and surgical complications, and the use of hydroxyurea, a chemotherapeutic agent. esearch is one key to keeping such statistics headed in the right direction. Over the past nine years, the Georgia Sickle Cell Center has initiated clinical studies of analgesic agents for acute pain, transfusion therapy for neurologic and surgical complications, and the use of hydroxyurea, a chemotherapeutic agent.
The new NIH grant provides the opportunity to greatly expand basic science and clinical studies of sickle cell disease at Emory. The grant covers research on ways to prevent sickle cell adhesion to the blood vessel walls, it focuses on problems of blood clotting and organ failure, and it also helps researchers explore the option of bone marrow transplant for patients who may benefit from the procedure. Much of the research on cell adhesion is done in collaboration with the Georgia Institute of Technology, where engineers Timothy Wick and Robert Nerem use an in vitro flow model of the blood vessel to study how sickle cells interact with endothelial cells lining the vessel wall. From an engineering perspective, the way sickle cells affect the circulation goes beyond biology to matters of force, flow rate, and shear stress. The engineered flow model allows Drs. Nerem and Wick to study various factors that may collaborate to impede blood flow. "In addition to plasma proteins, viruses, and other factors, flow and shear stress in a vessel can up- or down-regulate receptors that trigger adhesion," says Dr. Wick. "Sickle cells colliding with endothelial cells, not necessarily adhering to them, also may produce effects," says Dr. Nerem, "ranging from a change in cell morphology to a change in the gene expression level. We know, for example, that sickle cell patients have a high predilection for certain types of vascular disease." "Although we are a long way from helping regulate adhesion in patients, the hope is eventually to block or reverse it, at least temporarily, in people who sense they are just entering a pain crisis," Dr. Wick says. Adhesion of sickle cells also plays a major role in acute chest syndrome - a sudden and severe inflammation of the lungs and a common manifestation of sickle cell disease. "When we treat this syndrome, we are not really sure what we're treating," says Dr. Iris Buchanan, former director of Emory's pediatric sickle cell program. "It can initially look like mild pneumonia and then suddenly distribute over the entire lung." Hematologist Andreas Huber is investigating ways in which sickle cells stimulate the endothelium in the pulmonary vasculature to express adhesion receptors. This process in turn activates leukocytes, which cause inflammation and damage to the lungs. "We hope to determine the etiology of acute chest syndrome and then develop preventive measures and treatments," he says. Dr. Eckman and his colleagues believe that once cells begin to sickle and damage endothelial cells, they release factors which activate platelets in the blood, leading to clotting. Drs. Eckman, Laurence Harker, and Aaron Tomer are studying the thrombus-promoting properties of sickle cells during pain crises and the effect of giving patients dietary omega-3 fatty acids (fish oil), which have been shown in non-human primates to stop thrombus formation at sites of vascular injury. As patients with sickle cell disease live longer due to early and careful management, physicians must deal with problems resulting from chronic organ damage. Renal failure is becoming an important cause of death in older sickle cell patients. Early on, patients with renal problems have protein in the urine, which later progresses to a more severe decrease in renal function, sometimes requiring dialysis. Drs. Antonio Guasch and William Mitch are investigating how the glomeruli in the kidneys of sickle cell patients filter different types of protein. By determining precisely how the glomerulus is damaged, they hope to learn how to recognize signs of early kidney disease so that therapies can be designed to arrest or slow the loss of kidney function. |
 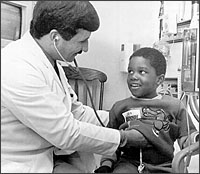 Five-year-old Seye Arise is the first patient at Emory to receive a bone marrow transplant for sickle cell disease. Before the procedure, which was done by Dr. Roger Vega, Seye often required hospitalization and had suffered a mild stroke, an indicator of poor prognosis for the future without a transplant.  For children having trouble adapting to their disease, Emory psychologist Nadine Kaslow uses intervention techniques to teach them and their families about sickle cell anemia and effective ways of coping with it. |
 ne of the newest alternatives for treating selective cases of sickle cell anemia is bone marrow transplantation. Emory's first patient to receive such a transplant was 5-year-old Seye Arise, who underwent the procedure in December. He received donor marrow from his older brother, who has the sickle cell trait but not the disease. ne of the newest alternatives for treating selective cases of sickle cell anemia is bone marrow transplantation. Emory's first patient to receive such a transplant was 5-year-old Seye Arise, who underwent the procedure in December. He received donor marrow from his older brother, who has the sickle cell trait but not the disease.
Born at Grady Hospital, Seye was diagnosed at birth with homozygous sickle cell anemia, the most common form of the disease. As a regular patient in the Georgia Sickle Cell Center, Seye has been taking prophylactic penicillin and folic acid since he was born. During his first year of life, he had no unusual symptoms. Soon after his first birthday, however, his hand and foot began to swell, signaling the beginning of one of seven major painful crises during the past four years. Seye also has experienced unexplained fevers, pneumonia, and abdominal swelling. A mild stroke in July led his physicians to recommend that he have a bone marrow transplant. As part of the NIH comprehensive sickle cell grant, Emory offers bone marrow transplantation as an option for children up to age 16 with sickle cell anemia whose disease is uncontrolled by conventional forms of supportive care and who have a poor prognosis for achieving a good quality of life into adulthood. Emory hematologists expect to perform transplants in six to 12 children each year for at least five years, for a total of 30 to 60 children. Emory is a member of the Seattle Bone Marrow Transplant Group - an association of eight centers nationwide exploring transplants in children with sickle cell anemia. The group's criteria for the initial transplants are stringent: All transplants will be allogeneic, using an HLA-matched full sibling with either normal hemoglobin or the sickle cell trait. Transplant candidates must have homozygous sickle cell disease and must have suffered a stroke or have severe visual impairment, progressive lung disease, chronic debilitating pain, or kidney damage. Organ damage must be early and reversible. The group predicts a greater than 90% event-free survival, based on transplant results in patients with thalassemia and aplastic anemia. More than 200 European children with thalassemia (an inherited anemia prevalent among Mediterranean populations) have received bone marrow transplants, with a cure rate of 80% to 90%. Thus far, about two dozen children in this country and Europe have received bone marrow transplants for sickle cell anemia, with a cure rate of 70% to 80%. These cases have included patients with acute leukemia or other malignancies in addition to sickle cell disease. Some deaths were due to these malignancies, and some failures were due to transplants in patients whose disease was too far advanced. Before the transplant, patients receive chemotherapy with busulfan and cyclophosphamide to destroy stem cells and immunosuppress the bone marrow. Cyclosporine and methotrexate are given to prevent graft-versus-host disease. Possible complications, as with any such transplant, include graft rejection, toxicity from chemotherapy, hemorrhage, and life-threatening infection. "With bone marrow transplants, you have up-front risk with the hope of long-term gain and the chance to live free of the problems associated with sickle cell disease," says Dr. John Wingard, director of Emory's bone marrow transplant program. "Unfortunately, in this group of patients, when you avoid the risk, you have a very high likelihood of further problems, deterioration, and perhaps death." The transplants are being performed at Egleston Children's Hospital by Dr. Roger Vega, who also performs pediatric transplants for other hematologic malignancies. Although finding a homozygous, full-sibling donor is ideal, it is rare, he says. Like Seye Arise, most patients will receive donor marrow with the sickle cell trait. Seye Arise was a good candidate for bone marrow transplant because his recovery from the stroke was excellent, says Dr. Vega. "We are looking for patients to whom we can offer a long-lasting, healthy life." For Seye's mother, the decision to choose a bone marrow transplant for her son seemed clear cut. "He is too little to be having these strokes now," she says. "I know if the bone marrow transplant is successful he is not going to have the disease anymore." |
Life Under the Sickle | The Shape of Things To Come
Inside a Private World | A Leader in Good Time
Copyright © Emory University. All Rights Reserved.
Web version by Jaime Henriquez.